



-
- Posts: 34
- Joined: Wed Aug 15, 2007 8:55 pm
hi everybody, first post to this forum. I have a bunch of questions related to our 5890gc. we are chemical engineers, not analytical chemists, so we have a good understanding of the gc, but little practical experience. Were trying to get this up and running and need some pointers on what is 'normal'. We had an incident which might have damaged our column, need some help getting going.
a little background:
astm d6584 uses mstfa to derivatize fatty-acid methyl esters, glycerol, and mono,di, and triglycerides. The goal is determination of free and total glycerin in biodiesel. Here is some more info:
http://www.sigmaaldrich.com/supelco/bul ... 107943.pdf
We are using manual injection with cool-on-column injector, and a 5m guard column
the method recommends injector temp of 50C with presumably temperature tracking oven temp
the person who sold us the gc was highly critical of the method and recommended injection at 100C with a 40C/min injector zone temp ramp rate to 380C. I assume this was to assure complete volatilization of the solvent to prevent tailing and protect the column
the oven temperature ramps at the following rates
hold at 50C for 1 min
15C/min to 180
7C/min to 230
30C/min to 380
FID detector is operated at 380C
we are using helium for the carrier gas
we created our standard and stock solutions last week, and shot a few to attempt to establish our calibration. Of our 4 standards (glycerol, mono-, di-, and tri-olein), only monoolein came out with a decent correlation coefficient. glycerol was particularly hard to detect as it was obscured by the tail of the heptane peak.
We decided to re-run the injections the next day, but we ran into an 'EPPB system shutdown'. It took us a few days to figure out that this was likely due to the injector septa, which we replaced. in the meantime, there was a sample that got left in there with no He purge (EPC shut itself down), and i'm afraid it damaged our column.
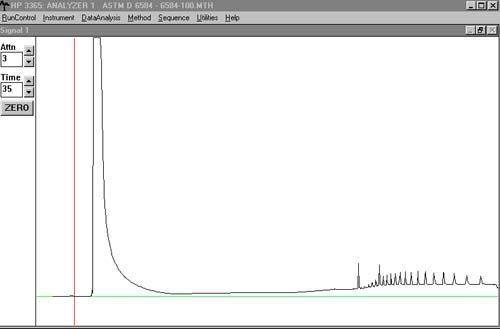
I made some new standard solutions this week, but everytime i inject a sample, we only get a few short broad peaks and the tailing of the heptane peak has gotten worse. I'm not seeing any of the internal standards or correlation standards. To try to figure out whats going on, I did a blank run:
here is a run with internal standards:
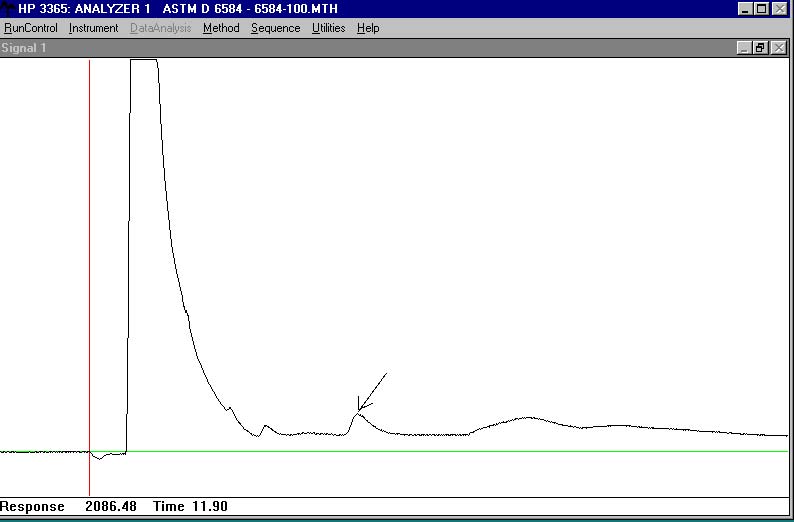
note that the attenuation was set to zero on this, i put a marker in to indicate the time and peak height at that point.
I'm not sure if this is due to poor column resolution due to contamination or maybe our mstfa got deactivated. (I purged the mstfa bottle with helium the best i could and kept it in the refrigerator all week. While not a perfect purge, i would expect it to keep longer than a week under those conditions, and at least show some activity.)
so in summary i have 4 possible thoughts here:
1. column/injector is badly contaminated, thus little to no resolution
2. the stock solutions i made last week (glycerol/monoolein/diolein/triolein/butanetriol/tricaprin, all in pyridine) have broken down and are unusable (these were stored in septa vials in the freezer. while not perfectly purged, i expect some resolution, so i'm gonna go out on a limb and say that #2 is less likely
3. MSTFA got de-activated, despite my best efforts
4. some combination of #1 and #3
I have a bunch more questions, but for now, if i could get some assistance in cleaning injector, and/or baking/trimming/saving my column and somehow verifying i have useful standards, that would be a great starting point.
thanks in advance!
a little background:
astm d6584 uses mstfa to derivatize fatty-acid methyl esters, glycerol, and mono,di, and triglycerides. The goal is determination of free and total glycerin in biodiesel. Here is some more info:
http://www.sigmaaldrich.com/supelco/bul ... 107943.pdf
We are using manual injection with cool-on-column injector, and a 5m guard column
the method recommends injector temp of 50C with presumably temperature tracking oven temp
the person who sold us the gc was highly critical of the method and recommended injection at 100C with a 40C/min injector zone temp ramp rate to 380C. I assume this was to assure complete volatilization of the solvent to prevent tailing and protect the column
the oven temperature ramps at the following rates
hold at 50C for 1 min
15C/min to 180
7C/min to 230
30C/min to 380
FID detector is operated at 380C
we are using helium for the carrier gas
we created our standard and stock solutions last week, and shot a few to attempt to establish our calibration. Of our 4 standards (glycerol, mono-, di-, and tri-olein), only monoolein came out with a decent correlation coefficient. glycerol was particularly hard to detect as it was obscured by the tail of the heptane peak.
We decided to re-run the injections the next day, but we ran into an 'EPPB system shutdown'. It took us a few days to figure out that this was likely due to the injector septa, which we replaced. in the meantime, there was a sample that got left in there with no He purge (EPC shut itself down), and i'm afraid it damaged our column.
I made some new standard solutions this week, but everytime i inject a sample, we only get a few short broad peaks and the tailing of the heptane peak has gotten worse. I'm not seeing any of the internal standards or correlation standards. To try to figure out whats going on, I did a blank run:
here is a run with internal standards:
note that the attenuation was set to zero on this, i put a marker in to indicate the time and peak height at that point.
I'm not sure if this is due to poor column resolution due to contamination or maybe our mstfa got deactivated. (I purged the mstfa bottle with helium the best i could and kept it in the refrigerator all week. While not a perfect purge, i would expect it to keep longer than a week under those conditions, and at least show some activity.)
so in summary i have 4 possible thoughts here:
1. column/injector is badly contaminated, thus little to no resolution
2. the stock solutions i made last week (glycerol/monoolein/diolein/triolein/butanetriol/tricaprin, all in pyridine) have broken down and are unusable (these were stored in septa vials in the freezer. while not perfectly purged, i expect some resolution, so i'm gonna go out on a limb and say that #2 is less likely
3. MSTFA got de-activated, despite my best efforts
4. some combination of #1 and #3
I have a bunch more questions, but for now, if i could get some assistance in cleaning injector, and/or baking/trimming/saving my column and somehow verifying i have useful standards, that would be a great starting point.
thanks in advance!

