



-
- Posts: 40
- Joined: Sat Jan 18, 2014 12:34 am
Hi everyone. So I've been trying method development to analyze a mixture of several fatty carboxylic acid derivatives. I am using a Waters Symmetry C18 column, 3x100mm, 3.5um, 100A at a flow rate of 0.5 ml/min, column temp 40C. My HPLC system is a Varian Prostar 335 DAD with 2 210 solvent pumps and a 410 autosampler with 100uL sample loop.
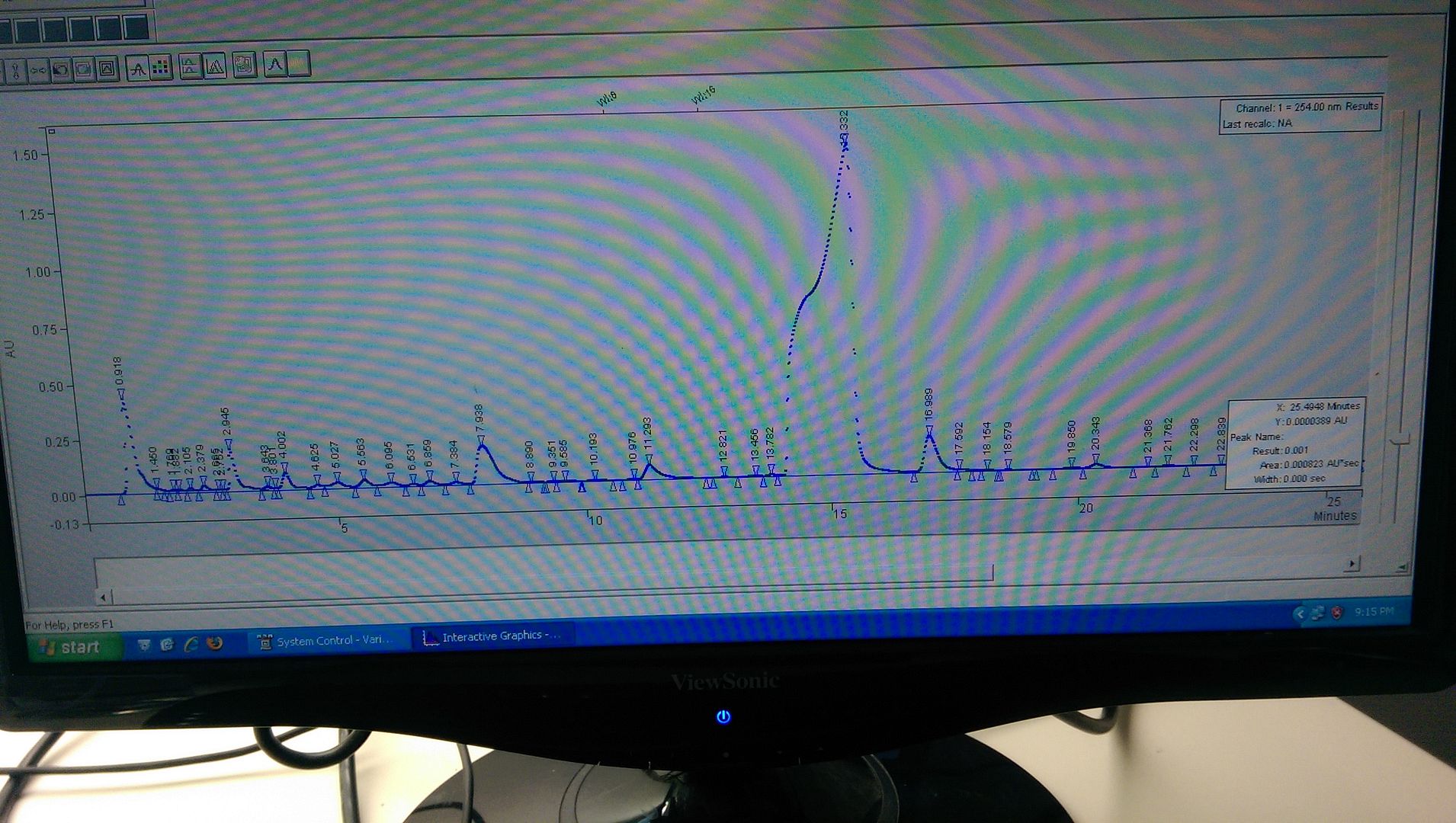
Initially, I tried using plain unbuffered MeOH:H2O. From 70:30 MeOH:H20 to 95:5 MeOH:H2O over 25min. The peak shape of the main component looked nice, however there was an uneven baseline due to gradient drift and several other components did not resolve well. Here is how it looked...

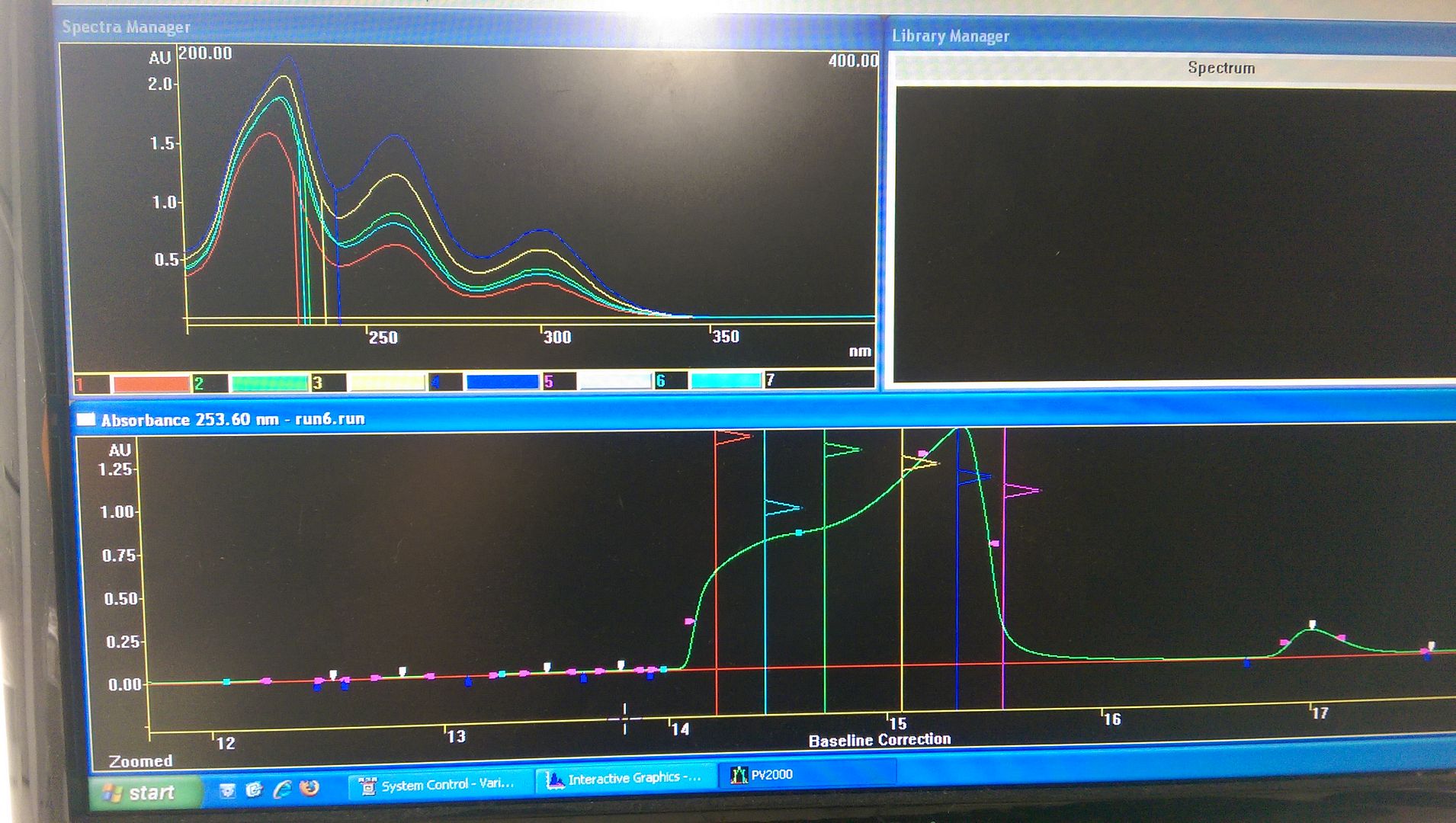
After reading up on some literature, I changed the solvent system to 35mM ammonium formate at pH 5.5. This time the baseline was beautiful, and most of the peaks gave sharp and great definition (some tailing). However the main component now shoulders...
Conclusions I've drawn...
1. Since it is the only peak that shoulders, it is probably not a frit or column guard problem.
2. I adjusted injection volume and concentration assuming overload, but there was not change in peak shape.
3. The injection solvent the sample is dissolved in matches the mobile phase concentration (70:30 MeOH:H2O - but it is not buffered, should it be?).
4. It most likely isn't a band broadening issue, as other peaks would be affected, and the problem occurred after a change in mobile phase.
5. The most obvious reason would be a co-eluted peak, however the purity factor on the DAD revealed the peak was pure, and scanning the UV of the peak across its whole width showed a similar absorbance...
Could I assume the change in peak shape is likely a pH issue? What would you recommend I do next, change buffer concentration and pH (should I go lower for a carboxylic acid?) Try a different column? Any advice would be helpful.
Edit: The buffer solution I made was 3 days old before I had a chance to use it, could this have also been an issue?
Initially, I tried using plain unbuffered MeOH:H2O. From 70:30 MeOH:H20 to 95:5 MeOH:H2O over 25min. The peak shape of the main component looked nice, however there was an uneven baseline due to gradient drift and several other components did not resolve well. Here is how it looked...

After reading up on some literature, I changed the solvent system to 35mM ammonium formate at pH 5.5. This time the baseline was beautiful, and most of the peaks gave sharp and great definition (some tailing). However the main component now shoulders...

Conclusions I've drawn...
1. Since it is the only peak that shoulders, it is probably not a frit or column guard problem.
2. I adjusted injection volume and concentration assuming overload, but there was not change in peak shape.
3. The injection solvent the sample is dissolved in matches the mobile phase concentration (70:30 MeOH:H2O - but it is not buffered, should it be?).
4. It most likely isn't a band broadening issue, as other peaks would be affected, and the problem occurred after a change in mobile phase.
5. The most obvious reason would be a co-eluted peak, however the purity factor on the DAD revealed the peak was pure, and scanning the UV of the peak across its whole width showed a similar absorbance...

Could I assume the change in peak shape is likely a pH issue? What would you recommend I do next, change buffer concentration and pH (should I go lower for a carboxylic acid?) Try a different column? Any advice would be helpful.
Edit: The buffer solution I made was 3 days old before I had a chance to use it, could this have also been an issue?


