-
- Posts: 9
- Joined: Wed May 25, 2011 9:48 pm
Hello All!  I am quite new here and to GC in general, so I hope you can bear with me. I will try to keep this post as concise as possible. I have organized it into 3 distinct sections: 1) my background, 2) what I am trying to accomplish with GC and 3) what problems I am facing.
I am quite new here and to GC in general, so I hope you can bear with me. I will try to keep this post as concise as possible. I have organized it into 3 distinct sections: 1) my background, 2) what I am trying to accomplish with GC and 3) what problems I am facing.
1) My Background: I am a graduate student in the field of mechanical engineering. My work is in experimental combustion. As a mechanical engineer, my chemistry is dismal at best, but I try my best to keep up with it when required (like now). I have read the first several chapters of Basic Gas Chromatography by Mcnair so I have a general (and qualitative) understanding of the underlying principles of GC.
I am using a Varian GC 3800 with Star Workstation 6.2 for software in order to test gas samples. The method that I am using is one that was developed by a Varian technician who gave on-sight classes before my arrival in this lab.
2) What I am trying to accomplish: This should be simple, I believe. I have tank that contains a known mixture composition of N2/CO2 (86/14). I would like to simply give this gas to the GC and calibrate it such that when I run an analysis, it can i) compute the correct composition when I give it another sample from the N2/CO2 mixture (i.e. it tells me that the mixture is 86/14) and ii) compute the percent composition of nitrogen if I give it a sample purified air (i.e. it tells me that the sample is 79% N2 and there are some other unknown constituents too).
I use a miniature regulator to monitor the pressure of the incoming sample gas at the inlet and a rotameter to monitor the flow at the outlet. I have included at snapshot of the setup below.
3) The problems I am facing: Remember that I am not even qualified to call myself a 'novice', so don't hesitate to assume that I am just plain dumb and state the obvious.
The way I have been instructed to do this (by someone who is only slightly more qualified than me) is to first calibrate the method by injecting a sample and taking note of the retention times. I then put these retention times into the 'peak table' of the method along with the gas names and their respective percentages. I then run the method as 'type calibration'. I then run the method again as a 'type analysis' and the result is the following image:
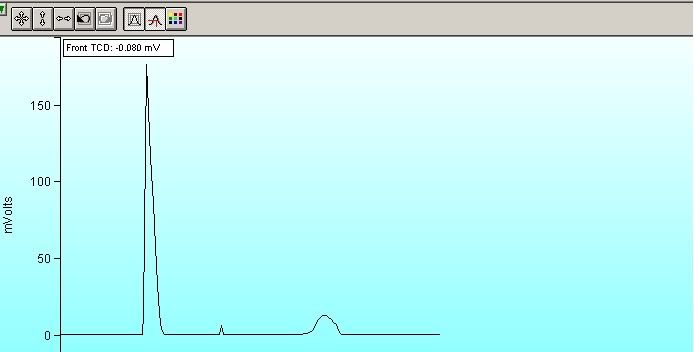
As you can see, there is a small peak that should not be there (I think). The N2 is on the left and the CO2 to the right. I have run this method about 7-8 times in an attempt to 'flush out' whatever that peak is. The area counts for this peaks have dropped with each run but they are taking longer and longer to reduce now (we are around 3500 counts now). Moreover, my results are a bit off. It is reporting ~85% N2 and ~15% CO2 instead of 86/14 and I think that the stray peak is what is causing this.
So my major questions are:
1) Does my procedure for calibration seem reasonable? And more importantly, when I "take not of the retention times" I have simply been using the figure generated and putting the it in 'cursor mode' and hovering over the peak and writing down the times. But what time should I be recording? The time at the highest point of the peak? Or the time that the peak starts to form? Also, is there a more precise way of doing this?
2) What are your thoughts on the stray peak? Are there any common reasons I should start trouble shooting?
Thanks so much for reading and please let me know if there is more information you need from me.
Image of gas inlet/outlet

1) My Background: I am a graduate student in the field of mechanical engineering. My work is in experimental combustion. As a mechanical engineer, my chemistry is dismal at best, but I try my best to keep up with it when required (like now). I have read the first several chapters of Basic Gas Chromatography by Mcnair so I have a general (and qualitative) understanding of the underlying principles of GC.
I am using a Varian GC 3800 with Star Workstation 6.2 for software in order to test gas samples. The method that I am using is one that was developed by a Varian technician who gave on-sight classes before my arrival in this lab.
2) What I am trying to accomplish: This should be simple, I believe. I have tank that contains a known mixture composition of N2/CO2 (86/14). I would like to simply give this gas to the GC and calibrate it such that when I run an analysis, it can i) compute the correct composition when I give it another sample from the N2/CO2 mixture (i.e. it tells me that the mixture is 86/14) and ii) compute the percent composition of nitrogen if I give it a sample purified air (i.e. it tells me that the sample is 79% N2 and there are some other unknown constituents too).
I use a miniature regulator to monitor the pressure of the incoming sample gas at the inlet and a rotameter to monitor the flow at the outlet. I have included at snapshot of the setup below.
3) The problems I am facing: Remember that I am not even qualified to call myself a 'novice', so don't hesitate to assume that I am just plain dumb and state the obvious.
The way I have been instructed to do this (by someone who is only slightly more qualified than me) is to first calibrate the method by injecting a sample and taking note of the retention times. I then put these retention times into the 'peak table' of the method along with the gas names and their respective percentages. I then run the method as 'type calibration'. I then run the method again as a 'type analysis' and the result is the following image:
As you can see, there is a small peak that should not be there (I think). The N2 is on the left and the CO2 to the right. I have run this method about 7-8 times in an attempt to 'flush out' whatever that peak is. The area counts for this peaks have dropped with each run but they are taking longer and longer to reduce now (we are around 3500 counts now). Moreover, my results are a bit off. It is reporting ~85% N2 and ~15% CO2 instead of 86/14 and I think that the stray peak is what is causing this.
So my major questions are:
1) Does my procedure for calibration seem reasonable? And more importantly, when I "take not of the retention times" I have simply been using the figure generated and putting the it in 'cursor mode' and hovering over the peak and writing down the times. But what time should I be recording? The time at the highest point of the peak? Or the time that the peak starts to form? Also, is there a more precise way of doing this?
2) What are your thoughts on the stray peak? Are there any common reasons I should start trouble shooting?
Thanks so much for reading and please let me know if there is more information you need from me.
Image of gas inlet/outlet