Page 1 of 2
Bad, Bad DMF
Posted: Mon Oct 29, 2007 4:03 pm
by Alchemist5
We routinely run residual solvents for numerous drug substances and excipients. For the majority of the time we use water or DMF as the diluent and use headspace (G1888) as the delivery system on Agilent 6890s.
There is one problem. On one of the instruments, when DMF is the diluent, we see breakdown products (probably dimethylamine or methylamine)....a peak near methanol that varies in size and has considerable tailing on 624 type columns.
I've changed liners, inlet seals, septa, but nothing seems to help. Would an active site somewhere in the G1888 be the source of this breakdown? It doesn't matter what temperature any of the system's components is, I still get this peak...and only on this system. Adding 1 ml 0.1N HCl to the HS vials eliminates my amine problem, but I can't always do this.
If it is the headspace sampler, what can I do to eliminate the active site. I'm assuming it's the needle, but can it be in the valve or transfer line?
Burt
Posted: Mon Oct 29, 2007 4:31 pm
by zokitano
Alchemist5,
Is it possible that you may have some "unknown" or "unexpected" compounds in the HS vial (from matrix etc.) that elute near methanol?
Is it possible maybe that with 0.1N HCl you convert this/these compound(s) into salts that are less or nonvolatile and therefore you don't observe them in the chromatogram when you'll add HCl in your vials?
Best regards
Posted: Mon Oct 29, 2007 4:34 pm
by chromatographer1
Hi Burt
We discussed this once before. I believe it is not an active site in your analyzer or GC that causes this problem. Remember it can be fixed by adding some acid to the solution before it is heated. The amine is there or is immediately created during the sample heating.
This impurity appears in the DMF because it breaks down in its bulk storage bottle and usually because there was some residual water in it to begin with or it picked up some water after being opened.
You might redistill your DMF and see if that helps. It that is not possible perhaps passing it through an ion exchange resin might help, but I doubt it.
Of course, if you add water to DMF and heat it in a headspace vial you are likely to make this impurity in the process of reaching equilibrium.
You are fortunate to have such a great GC system that it is not absorbed and it elutes as a peak.
You might try putting some active column tubing before your analytical column and see if that will lay down this amine peak so it will not interfere with your methanol integration.
Maybe a short piece of FFAP or SP-1000 capillary might work. I know, eyes are rolling all throughout the forum, but sometimes a chemist 'gots to do' what a chemist 'gots to do' to get it done.
I hope this is helpful if not amusing. Good luck,
Rod
Posted: Mon Oct 29, 2007 4:44 pm
by chromatographer1
Burt,
Let me add that if you do not have fused silica lined SS tubing for your sample loop and transfer lines you should change them immediately.
Also you might use a 0.28mm ID piece of fused silica SS tubing cut to a taper instead of a needle if you are inserting your transfer line into your injection port through a septum.
You might even wish to use a piece of FSOT column as long as you can trim the tip before you install it into the injection port after piercing the septum.
good luck,
Rod
Posted: Mon Oct 29, 2007 5:00 pm
by Alchemist5
I know this sounds like a solvent problem, but I do not see this impurity on my other instruments with exactly the same set-up. I believe it is not my DMF...I have tried other brands and purities. I too thought from our previous discussions, Rod, that it was something else. Using 0.1N HCl worked wonders, but I feel now it's only a band-aid on the true problem.
I have done a manual injection of 1µl DMF into the system and see no impurity...even after heating the DMF in a headspace vial. Only when I use the headspace sampler do I see it. This leads me back to the DMF actively breaking down on some active site within the sampler.
Weird, eh? So, humoring me, is there a way to deactivate my sampler? I'm now wondering if at anytime sample/standard solution splashed up to the top of the headspace vials and contaminated the needle sampling the vapor....
Posted: Mon Oct 29, 2007 5:11 pm
by chromatographer1
You wrote:
"Weird, eh? So, humoring me, is there a way to deactivate my sampler?"
Yes, there is.
Do you have a fused silica lined sample loop or a fused silica lined transfer line in your Agilent G1888?
If not, replace them. Bare metal may catalyze formation of this amine peak.
But consider what is different with this analyzer from the other analyzers you have.
Do you have bare metal lines in your other analyzers?
If the others are bare metal change your G1888 to bare metal also. That way your G1888 may absorb your amine peak in similar manner.
Have you replaced or cleaned your needle assembly/sample loop/transfer line lately? If not maybe it is time to replace with NEW.
Here are some ideas. Please describe what is different with this unit from your other units. It may become clear what change is necessary to free you from this nasty little peak. Which brings up a point.....
Just how big is this peak in comparison to a known amount of methanol for example? Can you tell us ?
Thanks and best wishes,
Rod
Posted: Mon Oct 29, 2007 5:17 pm
by Alchemist5
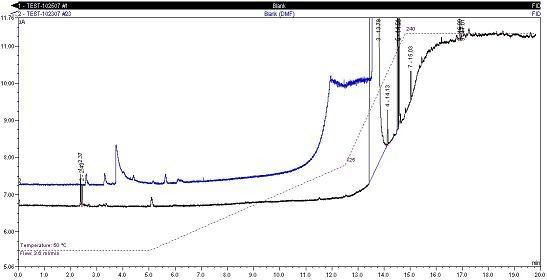
Here is an overlay of my manual injection (black) vs. the headspace injection (blue). Notice the 'peak' of concern at ~3.8 min and the large difference before the actual DMF peak (13.8 min). The early eluting peak is independent of DMF brand or purity, but dependent on introductory method.
These systems are < 6mo. old and all have the same new G1888 components. The only difference is that this system ran a few more headspace validations than the other 3 systems. I'm not sure how to clean the needle and will consult Agilent.
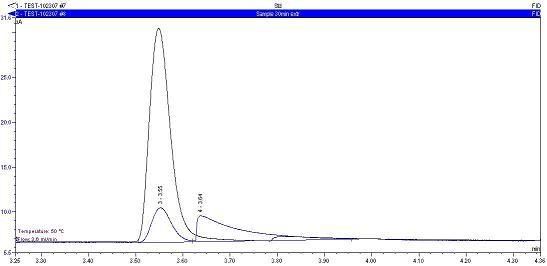
Peak in comparison the the methanol peak. This is an overlay of sample solution and a standard. The sample methanol peak (3.55 min) is much more affected by this peak.
Posted: Mon Oct 29, 2007 5:38 pm
by chromatographer1
Burt,
What kind of tubing does your G1888 have for sample loops and transfer lines?
Rod
Posted: Mon Oct 29, 2007 5:48 pm
by Alchemist5
Rod,
All I can find from Agilent is "A deactivated sampling path, from sampling needle to transfer line, is standard".
I was also directed to this. This describes a cleaning, not a deactivating procedure. All I can do is try it.
1. Set the carousel temperature to 60 degrees C and set the loop and transfer line temperatures to Off.
2. Once the temperatures of the loop and transfer line have fallen below 60 degrees C, shut off the gas to the Headspace Sampler and remove the vent line tubing at the outlet.
3 .Insert an empty vial in the carousel.
4. Raise the vial manually.
5 .Load about 6ml of purified water into a disposable syringe and inject from the HSS vent line.
6. Lower the vial and then remove. Repeat steps 3 to 5 (injecting two or three times will clean adequately).
7. Set the empty vial again and raise, then use the disposable syringe to inject enough air to flush out the water.
8. Insert the end of the transfer line into a beaker or similar and use the disposable syringe to inject about 10ml of water from the HSS gas inlet, followed by about 10ml of methanol.
9. Use the disposable syringe to inject enough air to flush out the water.
10. Put back the piping and turn on the gas. Next, switch the sample vial to Load-INJ several tens of times to blow out all of the water and methanol from the sample valve and similar.
Posted: Mon Oct 29, 2007 6:21 pm
by chromatographer1
Burt,
I am afraid I am at a point where I cannot make any further comments about the Agilent G1888 on this public forum. I do not know enough details specifically to be productive in correcting this issue.
The only help I can offer you is that perhaps you can find a way to flatten this amine peak in your chromatogram without affecting your other peaks.
Of course, that might require changing your column configuration which is probably something you are not permitted to do.
I do know that certain compounds (APIs) can react with the DMF and give you this interference, especially when there is water present in the API or in the DMF. Adding an organic acid (benzoic, acetic, etc) may help you overcome this issue.
Feel free to email me for private discussions if you are permitted to do so.
best wishes,
Rodney George
Senior Research and Development Scientist
Gas Separations Research
Supelco
595 North Harrison Road
Bellefonte, PA 16823
814-359-5737 voice
814-359-5459 fax
rodney.george@sial.com
Posted: Mon Oct 29, 2007 6:31 pm
by Alchemist5
Thanks Rod for your always welcome help. I will update if any changes occur.
Burt
Posted: Tue Oct 30, 2007 8:11 pm
by GoldLeader
Just a quick follow up question for this thread, You mentioned adding 0.1N HCl to DMF. How was this prepared? (i.e., 5 mL 0.1N HCl per L of DMF?...etc.) I ask because I sometimes see similar chromatography when using DMF diluent and I'm willing to try this approach. Also, I recall that DMF and HCl are not exactly compatible?
Thanks for any information.
Jeff
Posted: Tue Oct 30, 2007 10:49 pm
by chromatographer1
Goldleader,
In June of this year, in posting about a headspace artifact, Burt (Alchemist5) stated that he used 0.001N HCl added to the DMAc to decrease this amine peak. I had advised him to acidify his solution. DMAc acts in the same manner as DMF.
I wrote in explanation:
"When dimethylacetamide is heated with water over a length of time usually taken by most published methods a reaction takes place and methylamine(?) and dimethylamine(?) are formed and possibly other artifacts or impurities in the DMA are seen to elute.
Other reactions can take place depending upon the matrix and other factors.
This methylamine peak elutes early, often near methanol on many columns.
The peak near DMA is DMF or MMA and should elute just before DMA, this MMA is a normal small impurity found in most lots of DMA.
What can happen? If your flow path is reactive rather than inert (this is all relatively speaking now) the MeAmine or DiMeAmine can be absorbed rather than elute as a distinct peak. A well used system will often not elute the peak although other alcohols, ketones, amides, etc will elute without a noticeable problem.
The HCl you added makes an amine salt of the MeAmine or diMeAmine and inhibits its volatility so it never get to the flow path or at least is suppressed in concentration.
BTW, I found that if one uses smaller samples the LOD for most analytes does DOWN, not up, which is counterintuitive, but true. I never use more than 100µL of dissolved sample to do headspace analysis and am able to achieve a reproducible equilibrium for most residual solvents in 6 minutes of heating. Alcohols like i-BuOH or IPA will take 8 to 10 minutes to equilibrate to a near maximum level. This greatly reduces the artifacts that can be formed.
If you read my short two page HS article published in the Journal of Analytical Chemistry in 1997 you will see that I stated there:
"The dimethylacetamide produces artifactual peaks which coelute with methanol and dichloromethane and are adjacent to hexane, isobutanol, and dioxane. "
I only heated my samples for 6 minutes and was able to see and separate 1ppm of 18 residual solvents (including dioxane) using only 1mg of sample dissolved into 25µL of water (with a small amount of DMA).
I used the Tekmar 7000 modified with fused silica coated components, of which I was a beta tester. "
best wishes,
Rod
Posted: Wed Oct 31, 2007 5:51 pm
by Alchemist5
Hi Guys,
Just to follow up on the interfering peaks from breakdown products of DMA/DMF during heated headspace sampling, I want to mention that the observation of these peaks seems to be column dependent.
I had written that I was using a XX-624 column (6% Cyanopropylphenyl - 94% dimethylpolysiloxane), but in reality I was using a XX-5 (5% Phenyl - 95% methylpolysiloxane, 60m x 0.25mm x 1.4µm). With the latter, I observed amine elution near methanol RT and the DMA/DMF peak itself. No matter what parameter I altered of the system (temp, time, etc) I observed these peaks. I new they were amines, because amines do not deal well with this stationary phase, as they tend to have peaks that change RT, shape and size from run to run.
Adding 0.001N HCl to the headspace vial (I did this by adding 5mL of the DMA/DMF solution to be tested and then 1mL of 0.001N HCl) eliminating these peaks by converting them to non-volatile ionic species as Rod described. This I felt was only a band-aid on the situation, because I couldn't add HCl in every situation (the drug substance or method wouldn't allow for it). So, I tried to seek out an answer for this situation.
Was it the solvent itself? Or was it the GC or headspace sampler breaking it down? I changed all the consumable parts I could without any success. Manually injecting these solvent showed no amine compounds, so it had to be happening during the heating process or from the headspace sampler. Shorter incubation times did not help either and from what I could see, the headspace sampler (Agilent G1888) had all deactivated lines.
In a last ditch effort I tried a different column (the 624) on a developmental project where I could play with my hardware. I veered away from the XX-5 columns I was restricted to and found that I did not see these peaks. Now the question, are the amines being generated on the column (doubtful due to the liquid injection experiment) or during the heated incubation? If they are generated during the incubation, why don't I see them on the 624 column (30m x 0.53mm x 3.0µm)?
As for now, I may have averted my problem by column substitution. I can't believe it would be the headspace sampler lines, because these instruments are less than 6 months old and have not been abused with nasty samples like in my old days of environmental analyses.
Burt
Posted: Wed Oct 31, 2007 5:52 pm
by Alchemist5
Hi Guys,
Just to follow up on the interfering peaks from breakdown products of DMA/DMF during heated headspace sampling, I want to mention that the observation of these peaks seems to be column dependent.
I had written that I was using a XX-624 column (6% Cyanopropylphenyl - 94% dimethylpolysiloxane), but in reality I was using a XX-5 (5% Phenyl - 95% methylpolysiloxane, 60m x 0.25mm x 1.4µm). With the latter, I observed amine elution near methanol RT and the DMA/DMF peak itself. No matter what parameter I altered of the system (temp, time, etc) I observed these peaks. I new they were amines, because amines do not deal well with this stationary phase, as they tend to have peaks that change RT, shape and size from run to run.
Adding 0.001N HCl to the headspace vial (I did this by adding 5mL of the DMA/DMF solution to be tested and then 1mL of 0.001N HCl) eliminating these peaks by converting them to non-volatile ionic species as Rod described. This I felt was only a band-aid on the situation, because I couldn't add HCl in every situation (the drug substance or method wouldn't allow for it). So, I tried to seek out an answer for this situation.
Was it the solvent itself? Or was it the GC or headspace sampler breaking it down? I changed all the consumable parts I could without any success. Manually injecting these solvent showed no amine compounds, so it had to be happening during the heating process or from the headspace sampler. Shorter incubation times did not help either and from what I could see, the headspace sampler (Agilent G1888) had all deactivated lines.
In a last ditch effort I tried a different column (the 624) on a developmental project where I could play with my hardware. I veered away from the XX-5 columns I was restricted to and found that I did not see these peaks. Now the question, are the amines being generated on the column (doubtful due to the liquid injection experiment) or during the heated incubation? If they are generated during the incubation, why don't I see them on the 624 column (30m x 0.53mm x 3.0µm)?
As for now, I may have averted my problem by column substitution. I can't believe it would be the headspace sampler lines, because these instruments are less than 6 months old and have not been abused with nasty samples like in my old days of environmental analyses.
Burt