Page 1 of 2
Method transfer problem
Posted: Tue May 22, 2007 8:59 am
by tryfan
Hi all
I'm new to the list so please excuse any mistakes I make.
I work for a new pharmaceutical company who have taken over an old line from a bigger multinational - no names!! My problem is that I am trying to recreate their HPLC (UV det) results with little success. There are two problems, firstly and biggest is that I consistantly get 1.5mg/g higher results than them (average is about 48mg/g), no matter what I try its always the same. I use the standard that they supply so there can be no difference there. If I measure it at 47.5 they get 46.0, for example.
The other thing - maybe connected - is that I get slightly different traces than them. I've done alll the normal stuff such as changed bulbs in the UV detector, changed pumps, detectors, intergration systems basically the whole system in stages and I still get the same trace. I've tried different concnetration eluants, new columns etc but to no avail, I still get my normal trace which is different to theirs.
I use their method which specifies everything except what colour labcoat to wear, and follow it to the letter. Except in the method they gave me it has a trace which is the same as what i get, not what they get now...... But chatting to their lab person nothing has changed since the method was wrote!
Its now getting to the stage where to meet their exact requirements we aim high so their "lower" readings meet their needs. Obviously the two problems could be related, but the difference in the extra traces would not account for the difference in the results.
I'm not too worried at this stage about who is right or wrong, I just want to match their results so our little company can send our product out without it being rejected

Any ideas, hints or hot mugs of tea and sympathy greatly appreciated.
Many Thanks
Tryfan
Posted: Tue May 22, 2007 9:13 am
by Peter Apps
I notice that the column is not on the list of things that you changed, so best guess has to be that their column has deteriorated over time and that you are using a new one. Depending on how the analysis is calibrated this could give exactly the effect you see.
Peter
Posted: Tue May 22, 2007 9:21 am
by tryfan
Hi Peter
thanks for the reply. I have changed the column, in fact I've used three or four new ones since this has started (they are all still in good working order but changed to try and match them). My columns are in pretty good nick, looked after, changed guard column regularly etc. I can't comment on their's but I'll try to find out, thanks for the tip.
Tryfan
Posted: Tue May 22, 2007 11:20 am
by JM
If you are using external standard for quantification, there should not be any difference other than normal analytical error associated with method. Are you sure you are using the right potency of standard?
Two different make HPLCs can give slightly different chromatograms nothing more. What you mean different trace? did you get some extra peaks? or the elution pattern is different?
JM
Posted: Tue May 22, 2007 12:11 pm
by tryfan
Thanks JM
Its definately the right potency standard, as i said its supplied by them, and used to calibrate my results (they use the same one too).
By different traces I mean that I get more seperation than they do. For example a major peak they get I manage to find a shoulder in front of and sometimes split it away (eluant dependant), its there on their method they gave me if you look closely but not on their current traces. There are a couple of small differences like that but they don't add up to much overall. I also get a few peaks they don't on their current trace (but did on their test trace they supplied) but these are not near the measured components so I'm not worried over these too much. Elution pattern is the same.
After asking, they last changed their column 4 or 5 months ago (they won't say exactly) but are still getting good plate counts, over 1300. I presume that all is well in that respect.
Thanks again
Tryfan
Posted: Tue May 22, 2007 12:23 pm
by Jackus
Hello tryfan
Is possible to see both chromatograms? What is major peak height?
Isn't detector in non-linear region?
Are the instruments same type (manufacturer, models)?
Lot of questions-sorry

Posted: Tue May 22, 2007 4:06 pm
by Alfred88
Dear tryfan:
Did you (and the other company) use identical samples for testing? If your samples are from batches made at your company, check the balances.
Also, do a null hypothesis: Muy1 = Muy2, and use a t-distribution with 95% confidence to see if the difference is statistically significant.
Good luck.
Alfred
Tryfan
Posted: Tue May 22, 2007 4:10 pm
by AdrianF
I suggest you get someone to make up several solutions of known concentration and for each lab to anaylse them blind. From the results you will know then which lab is giving the best result.
Posted: Tue May 22, 2007 6:53 pm
by Hollow
Hi tryfan
is your error of absolut or relative origin, I mean do you alway get 1.5 mg/g higher or always 103.2%?
If it's relative I would also think on the purity and/or assay of your external standard.
If the other company is correcting for this and you don't this would also lead to such a phenomenon. (Maybe you are correcting "only" for purity and the others are correcting for assay.)
Another tipp is to do an exchange of your test solutions (calibration and sample) and have them analyzied at both labs.
Posted: Wed May 23, 2007 12:46 pm
by tryfan
thanks all
some good ideas there. I've uploaded some traces for inspection. Mine is

whilst theirs is
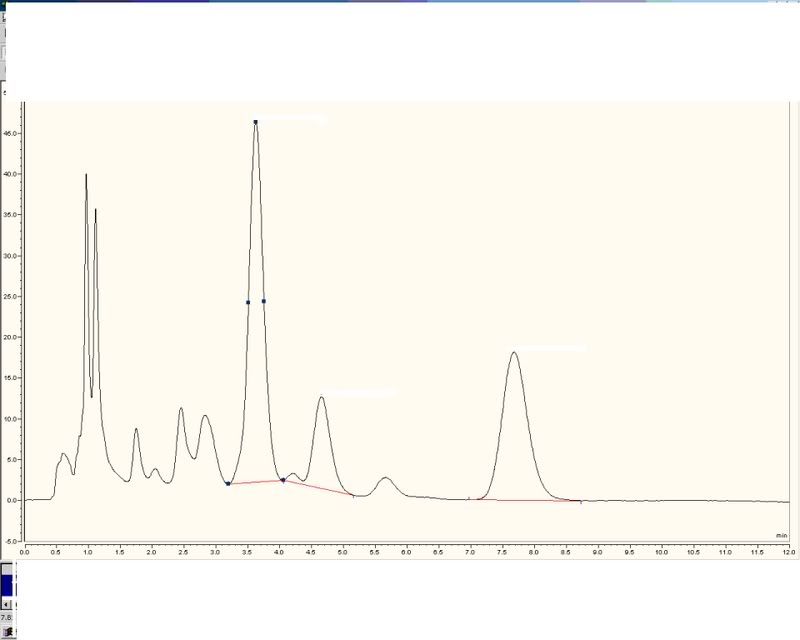
peaks 14, 15 and 23 are the important ones.
Hope this works.
Foefot to say, sometimes they dont use the small peak before peak 15, sometimes they do, but the overall result does not change much as its is so small.
Many thanks
Tryfan
Posted: Wed May 23, 2007 12:56 pm
by DR
The times are way off. Someone is not running at the correct flow rate. If the pressures are also way off, when running at the same flow rates (as confirmed by testing w/ volumetric glassware & stopwatches), someone's column is in dire need of replacement, or someone is not preparing their MP correctly.
If all of these things turn out to be the same, I would look at the calculations. Make sure that everyone is using the same purity & moisture corrections for the standards.
Chromatograms
Posted: Wed May 23, 2007 1:41 pm
by AdrianF
Apart from the retention times being different I am confused by your remark about using three peaks - are you measuring the three different components or obtaining one value by adding the three together in some way.
Posted: Wed May 23, 2007 1:55 pm
by tryfan
Thanks again
yes, the retention times happen to be diff in that example. I've been playing about with eluant strength to try and seperate the small peak, the second trace is more realistic day to day retention timewise. Even with same retention times results differ. The calculation combines all three peaks in the ratio 0.5, 0.1 and 0.4, giving a total active ingredient contained in the sample.
MP is an acetonitrile in 1% acteic acid mix, retention times vary with each MP made up only slightly, unless like above I'm trying different things.
Out of interest or ignorance, surely if the retention times were different, the areas under the peaks would still be comparable with others?
Tryfan
Posted: Wed May 23, 2007 2:02 pm
by AdrianF
Does the standard have the three peaks or just the main peak.
Posted: Wed May 23, 2007 2:27 pm
by tryfan
Hi Adrian
the standard has the three main peaks. its supplied by the customer as a known standard which their lab also uses to calibrate. Its basically the same as the samples but with a known amount of the three peaks.
Cheers