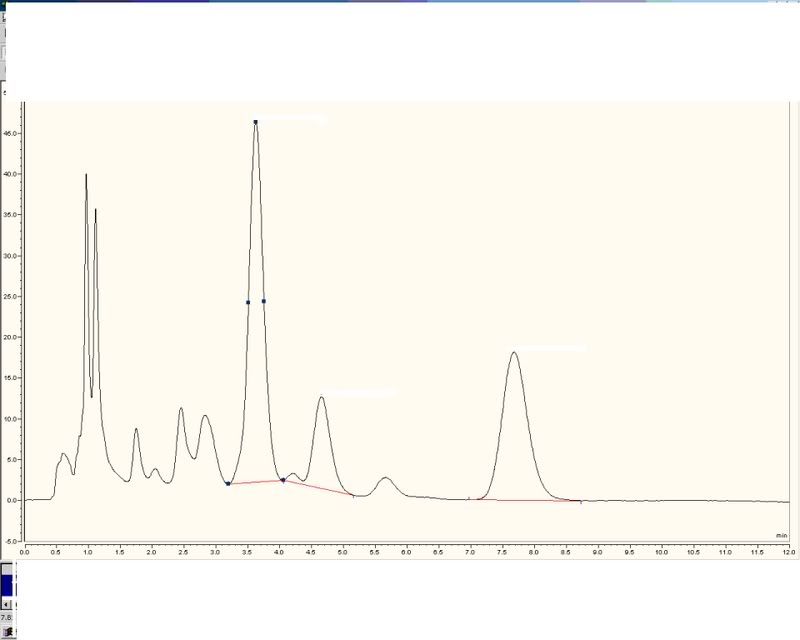
-
- Posts: 6
- Joined: Tue May 22, 2007 8:37 am
Hi all
I'm new to the list so please excuse any mistakes I make.
I work for a new pharmaceutical company who have taken over an old line from a bigger multinational - no names!! My problem is that I am trying to recreate their HPLC (UV det) results with little success. There are two problems, firstly and biggest is that I consistantly get 1.5mg/g higher results than them (average is about 48mg/g), no matter what I try its always the same. I use the standard that they supply so there can be no difference there. If I measure it at 47.5 they get 46.0, for example.
The other thing - maybe connected - is that I get slightly different traces than them. I've done alll the normal stuff such as changed bulbs in the UV detector, changed pumps, detectors, intergration systems basically the whole system in stages and I still get the same trace. I've tried different concnetration eluants, new columns etc but to no avail, I still get my normal trace which is different to theirs.
I use their method which specifies everything except what colour labcoat to wear, and follow it to the letter. Except in the method they gave me it has a trace which is the same as what i get, not what they get now...... But chatting to their lab person nothing has changed since the method was wrote!
Its now getting to the stage where to meet their exact requirements we aim high so their "lower" readings meet their needs. Obviously the two problems could be related, but the difference in the extra traces would not account for the difference in the results.
I'm not too worried at this stage about who is right or wrong, I just want to match their results so our little company can send our product out without it being rejected
Any ideas, hints or hot mugs of tea and sympathy greatly appreciated.
Many Thanks
Tryfan
I'm new to the list so please excuse any mistakes I make.
I work for a new pharmaceutical company who have taken over an old line from a bigger multinational - no names!! My problem is that I am trying to recreate their HPLC (UV det) results with little success. There are two problems, firstly and biggest is that I consistantly get 1.5mg/g higher results than them (average is about 48mg/g), no matter what I try its always the same. I use the standard that they supply so there can be no difference there. If I measure it at 47.5 they get 46.0, for example.
The other thing - maybe connected - is that I get slightly different traces than them. I've done alll the normal stuff such as changed bulbs in the UV detector, changed pumps, detectors, intergration systems basically the whole system in stages and I still get the same trace. I've tried different concnetration eluants, new columns etc but to no avail, I still get my normal trace which is different to theirs.
I use their method which specifies everything except what colour labcoat to wear, and follow it to the letter. Except in the method they gave me it has a trace which is the same as what i get, not what they get now...... But chatting to their lab person nothing has changed since the method was wrote!
Its now getting to the stage where to meet their exact requirements we aim high so their "lower" readings meet their needs. Obviously the two problems could be related, but the difference in the extra traces would not account for the difference in the results.
I'm not too worried at this stage about who is right or wrong, I just want to match their results so our little company can send our product out without it being rejected
Any ideas, hints or hot mugs of tea and sympathy greatly appreciated.
Many Thanks
Tryfan