Page 1 of 1
Odd peak shape with increased sample load
Posted: Thu Dec 11, 2014 2:40 pm
by DJ
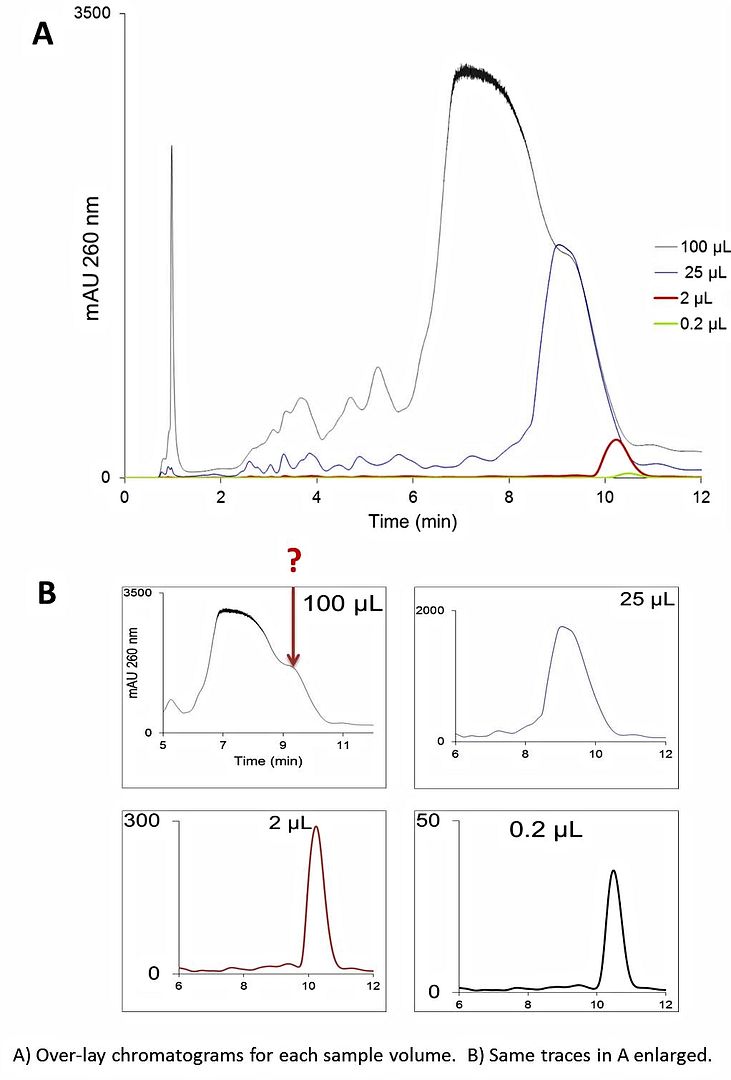
This is a loading study done with a crude 25 mer DNA hetero oligo (the initial purity of the crude oligo was ~ 73%). The majority of the impurities were n-1, n-2, failure sequences (which elute before the target sequence).
A= 0.1 M TEAA pH 7 + 2% MeCN
B= A + 20% MeCN
Temp: 60 C
Flow rate: 1 mL/min
Gradient: 5 to 15% B over 2 min, 15-30% B over 30 min.
Column: hybrid (inorganic/organic) ODS, 5 micron, 4.6 x 50 mm (advertised to have enhanced pH and temperature stability).
Prior to use, I evaluated column performance under the manufacturer's QA test conditions (in addition to several gradient separations of a polyd(T) 12-24 ladder, which is a more qualitative assessment of performance). The column is in great condition.
Why does the peak become distorted like this at the higher sample load?
Re: Odd peak shape with increased sample load
Posted: Thu Dec 11, 2014 2:52 pm
by H2Oh
Two initial things come to mind. I'm sure there are more answers though.
First is that you are simply overloading the column. There isn't enough interaction between your stationary phase and the analyte to retain it because all of the active sites of the stationary phase are already occupied and the excess sample isn't being retained well.
Secondly, if you have a large volume injection, the sample solvent replaces the mobile phase and creates a slug which is a different set of chromatographic conditions than your A and B streams. Your injection isn't all that large considering you are using 1 ml/minute and a 4.6 mm diameter column. It is something to keep in mind though. It's conceivable that the sample partitions out temporarily to a portion that is still under the injection conditions and one that has mixed with your mobile phase/buffer.
Re: Odd peak shape with increased sample load
Posted: Thu Dec 11, 2014 3:00 pm
by Consumer Products Guy
Looks overloaded to me as well.
I always try smaller volumes in development. My rule of thumb is that if peak shape improves with injecting less material or volume, then go with the reduced.
Re: Odd peak shape with increased sample load
Posted: Thu Dec 11, 2014 4:29 pm
by DJ
Two initial things come to mind. I'm sure there are more answers though.
First is that you are simply overloading the column. There isn't enough interaction between your stationary phase and the analyte to retain it because all of the active sites of the stationary phase are already occupied and the excess sample isn't being retained well.
Secondly, if you have a large volume injection, the sample solvent replaces the mobile phase and creates a slug which is a different set of chromatographic conditions than your A and B streams. Your injection isn't all that large considering you are using 1 ml/minute and a 4.6 mm diameter column. It is something to keep in mind though. It's conceivable that the sample partitions out temporarily to a portion that is still under the injection conditions and one that has mixed with your mobile phase/buffer.
The oligo was dissolved and loaded in buffer A.
If the solute band is excessively broad, could the front and rear portions of the peak be experiencing localized differences in the ratio of stationary phase: mobile phase ion pairing agent as well as % MeCN? I would imagine partition of ion pairing agent between the stationary and mobile phase to be sensitive to % MeCN. Higher % MeCN probably promotes partition into the mobile phase, while lower % MeCN promotes it. Could this be what is happening?
Re: Odd peak shape with increased sample load
Posted: Thu Dec 11, 2014 4:35 pm
by DJ
Looks overloaded to me as well.
I always try smaller volumes in development. My rule of thumb is that if peak shape improves with injecting less material or volume, then go with the reduced.
Yes, I agree.. However I'm evaluating performance of these columns for preparative applications.
Re: Odd peak shape with increased sample load
Posted: Fri Dec 12, 2014 8:32 am
by M_Farooq
Split peaks (or shoulders) are less common under column overload. Search for "Non linear chromatography". You chromatogram is just complicated by a shoulder, otherwise it is a classical tailing effect where the apparent peak retention time shifts earlier but the tails overlap. Another problem is that if the sample solvent is very different or in a stronger mobile phase one can see shoulders as well. How sure are you about that shoulder that is not an impurity?
If you are really interested in peak shapes then have a look at
https://www.elsevier.com/books/fundamen ... 2-370537-2
Re: Odd peak shape with increased sample load
Posted: Fri Dec 12, 2014 3:38 pm
by DJ
I'm all but certain the shoulder is not an impurity because I would expect it to be more and more apparent with decreasing sample load. Here, it's just the opposite: the deformed peak appears only under the highest sample load.
This sample has been analyzed by LC-MS. The target is 72-75% pure. There are many impurities, but all present at < 5% with the vast majority eluting earlier than the target.
I've purified this same crude oligo on two PS-DVB-based columns and the peak shapes were what you would expect for a "well behaved" sample under prep conditions.
Re: Odd peak shape with increased sample load
Posted: Fri Dec 12, 2014 3:58 pm
by Klaus I.
Since he have injected also a smaller volume of sample solution (we can observe this in the under part of the posting) a partly coeluting impurity would not be my first suggestion. This peak-shape can be observed quite often when we are overloading a column. Maybe this peak shape can be simply explained by a combination of volume-overloading and mass-overloading. But usually this problem occurs due to the solvent. This peak-shape is typical for to strong (to much organic) solvents but it is also typical for choosing a wrong pH.
Unfortunately I have no experience in this kind of chromatography, but I think it would be a good idea to make same calculation about the needed capacity of the used buffers for mobile phase and/or for the sample solvent.
Re: Odd peak shape with increased sample load
Posted: Tue Dec 16, 2014 1:13 pm
by DJ
Since he have injected also a smaller volume of sample solution (we can observe this in the under part of the posting) a partly coeluting impurity would not be my first suggestion. This peak-shape can be observed quite often when we are overloading a column. Maybe this peak shape can be simply explained by a combination of volume-overloading and mass-overloading. But usually this problem occurs due to the solvent. This peak-shape is typical for to strong (to much organic) solvents but it is also typical for choosing a wrong pH.
Unfortunately I have no experience in this kind of chromatography, but I think it would be a good idea to make same calculation about the needed capacity of the used buffers for mobile phase and/or for the sample solvent.
This is ion-pair RP-HPLC. The ion-pairing agent is 0.1 M triethylammonium acetate, pH 7. Neither acetate nor triethylamine are buffers in this pH range. However this method is widely used to purify oligonucleotides.
I agree the column is probably over-loaded. However, could someone explain (in handwaving) why a homogeneous species takes on THAT particular deviation in shape from the classic "shark fin" one sees under normal prep LC?
Re: Odd peak shape with increased sample load
Posted: Tue Dec 16, 2014 4:48 pm
by H2Oh
It may be that the ion pairing is incomplete or inconsistent. I've run ion pairing upon occasion and the MS clearly showed multiple ratios of ion pairing agent to analyte. I suspect that at lower concentrations/load, most of your sample is pairing nicely. At higher concentrations either you have some analyte that is not paired or not as completely pairs.
Let's say that each of your oligos wants to pair with five agents. At a relatively low concentration, there is enough pairing agent to bind all fully with five agents.
As you keep adding analyte with larger and larger injections, at some point you won't have enough pairing agent and some of your oligos may only have three ligands. These will behave similarly to the ones with five, but would show up as a separate peak. In your case, an overlapping separate peak.
You could test this hypothesis by pairing the analyte offline or upping the ion pairing agent and see if the peak morphs into the usual shark fin of an overloaded column.
That's my hunch.
Re: Odd peak shape with increased sample load
Posted: Tue Dec 16, 2014 4:53 pm
by H2Oh
...and based on your chromatograms, the overloaded samples show a peak earlier than the lower volume/concentration injections. This supports the idea of an incompletely paired analyte. Without as much ion pairing agent, the stationary phase has less interaction with the analyte and it elutes earlier.
Quick summary. The early peak is a poorly retained fraction of your oligo because of incomplete interaction with your ion pairing agent. Either due to too much sample, or too little ion pairing agent (or perhaps something else). It depends on your point of view.
Re: Odd peak shape with increased sample load
Posted: Wed Dec 17, 2014 2:26 pm
by DJ
Great insight. Thanks.
I repeated this separation yesterday with extensive equilibration with the ion pairing agent (MeCN content kept <5%) but got similar results.
What do you make of the observation that replication of this experiment on a different, PS-DVB-based column (same hardware and particle size as the XTerra- 4.6 x 50 mm, 5 micron) with the same gradient slope (but higher starting and ending %B, since oligos tend to be more well retained on PS-DVB vs XTerra column), there is no such deviation in peak shape under the highest sample load?
Could the PS-DVB column simply have a higher capacity for the ion-pairing agent?
Re: Odd peak shape with increased sample load
Posted: Wed Dec 17, 2014 6:54 pm
by tom jupille
Higher surface area? Possibly different secondary interactions (no silanols)?