



-
- Posts: 252
- Joined: Sat Nov 07, 2009 6:27 pm
This is a loading study done with a crude 25 mer DNA hetero oligo (the initial purity of the crude oligo was ~ 73%). The majority of the impurities were n-1, n-2, failure sequences (which elute before the target sequence).
A= 0.1 M TEAA pH 7 + 2% MeCN
B= A + 20% MeCN
Temp: 60 C
Flow rate: 1 mL/min
Gradient: 5 to 15% B over 2 min, 15-30% B over 30 min.
Column: hybrid (inorganic/organic) ODS, 5 micron, 4.6 x 50 mm (advertised to have enhanced pH and temperature stability).
Prior to use, I evaluated column performance under the manufacturer's QA test conditions (in addition to several gradient separations of a polyd(T) 12-24 ladder, which is a more qualitative assessment of performance). The column is in great condition.
Why does the peak become distorted like this at the higher sample load?
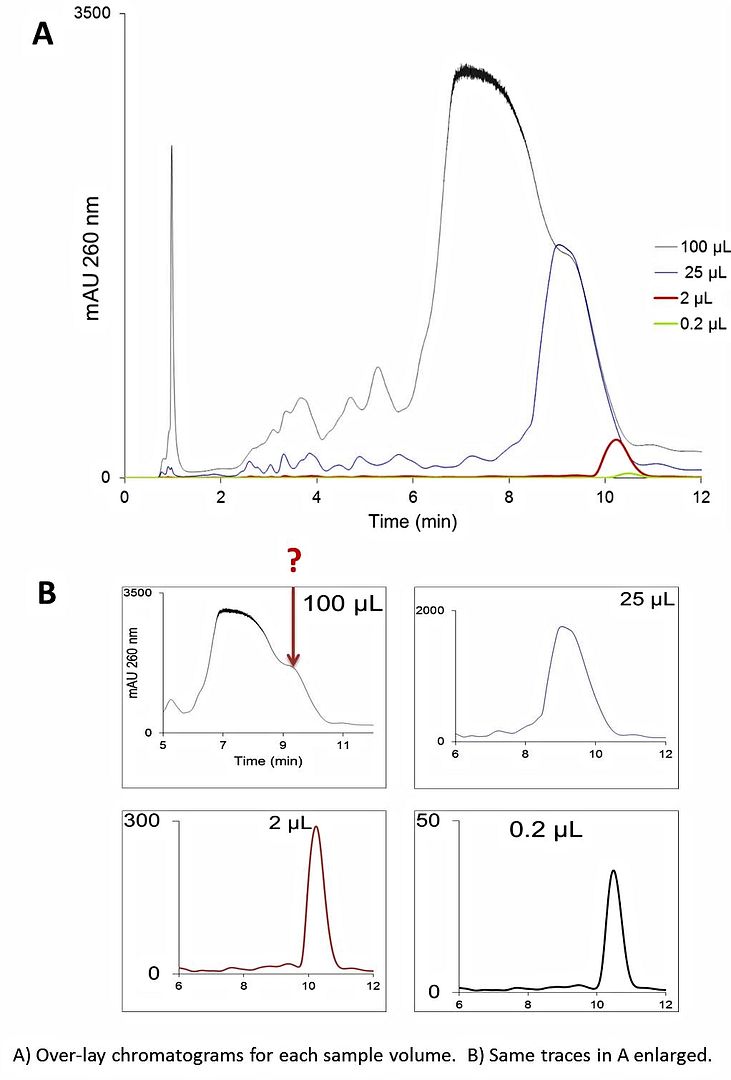
A= 0.1 M TEAA pH 7 + 2% MeCN
B= A + 20% MeCN
Temp: 60 C
Flow rate: 1 mL/min
Gradient: 5 to 15% B over 2 min, 15-30% B over 30 min.
Column: hybrid (inorganic/organic) ODS, 5 micron, 4.6 x 50 mm (advertised to have enhanced pH and temperature stability).
Prior to use, I evaluated column performance under the manufacturer's QA test conditions (in addition to several gradient separations of a polyd(T) 12-24 ladder, which is a more qualitative assessment of performance). The column is in great condition.
Why does the peak become distorted like this at the higher sample load?
