



-
- Posts: 45
- Joined: Tue Aug 09, 2011 9:16 am
Hello,
I just have two simple questions:
1. How to measure the area under the curve if you have two spikes that are not very well separated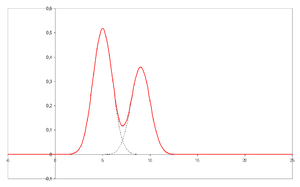 like in the image? Do you measure like usual or you separate the two spikes with a vertical line that goes down to the baseline and measure the area like that? What if the baseline is not really horizontal and you have noise on it, how do you measure then?
like in the image? Do you measure like usual or you separate the two spikes with a vertical line that goes down to the baseline and measure the area like that? What if the baseline is not really horizontal and you have noise on it, how do you measure then?
2. Is it possible to calculate LOD and LOQ in HPLC using the Mean + 3*Sigma and Mean + 10*Sigma, if yes what exactly can you measure in the baseline noise to get an area under the curve and where do you measure it?
Thank you
I just have two simple questions:
1. How to measure the area under the curve if you have two spikes that are not very well separated
like in the image? Do you measure like usual or you separate the two spikes with a vertical line that goes down to the baseline and measure the area like that? What if the baseline is not really horizontal and you have noise on it, how do you measure then?2. Is it possible to calculate LOD and LOQ in HPLC using the Mean + 3*Sigma and Mean + 10*Sigma, if yes what exactly can you measure in the baseline noise to get an area under the curve and where do you measure it?
Thank you
