Posted: Fri Aug 17, 2007 2:38 am
this is very confusing! i ran std3 right afterwards, and am starting to get peaks again, but its inconsistent:
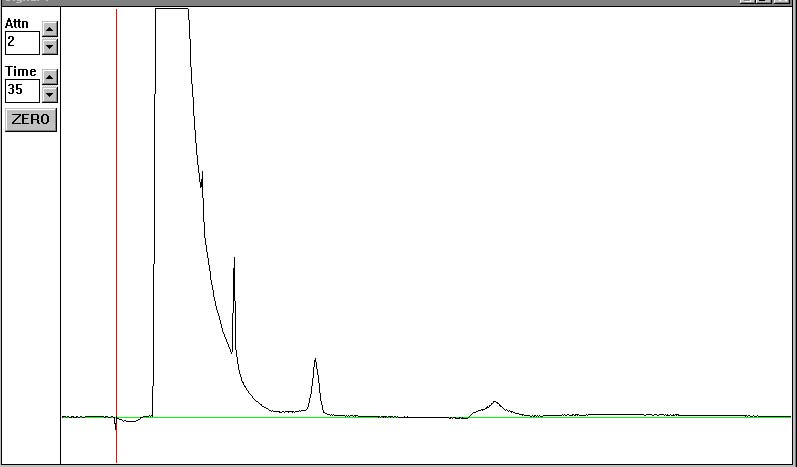
the y-axis is about 40000 in the following chromatographs
std3 - first run:
def seeing some peaks, but the solvent tail is still making it difficult.
=========================================
Area Percent Report
=========================================
Data File Name : C:\HPCHEM\1\DATA\NV-F0137.D
Operator : Page Number : 1
Instrument : ANALYZER Vial Number :
Sample Name : Injection Number :
Run Time Bar Code: Sequence Line :
Acquired on : 16 Aug 07 06:44 PM Instrument Method: 6584-100.MTH
Report Created on: 16 Aug 07 07:16 PM Analysis Method : 6584-100.MTH
Sig. 1 in C:\HPCHEM\1\DATA\NV-F0137.D
Pk# Ret Time Area Height Type Width Area %
|---|----------|--------------|--------------|----|---------|----------|
1 1.345 2606 165 BV 0.198 0.0003
2 1.567 86 17 VB 0.063 0.0000
3 1.746 7818 5836 BV 0.022 0.0008
4 1.949 8.99766E+008 1.31258E+008 VV 0.085 94.5063
5 2.004 5.0963E+007 5806131 VV 0.106 5.3529
6 4.094 962430 22965 VV 0.496 0.1011
7 5.645 230977 14254 VV 0.199 0.0243
8 9.458 41810 4701 VV 0.148 0.0044
9 9.525 19902 5259 VV 0.063 0.0021
10 9.541 59716 5182 VV 0.192 0.0063
11 17.090 1754 235 BV 0.093 0.0002
12 17.151 1039 265 VV 0.055 0.0001
13 17.214 621 238 VV 0.034 0.0001
14 17.328 1642 244 VV 0.087 0.0002
15 17.358 198 202 VV 0.016 0.0000
16 17.458 1061 170 VV 0.103 0.0001
17 17.704 1371 94 VV 0.184 0.0001
18 17.765 429 139 VV 0.040 0.0000
19 17.805 328 181 VV 0.030 0.0000
20 17.980 2333 350 VV 0.083 0.0002
21 18.043 1287 406 VV 0.053 0.0001
22 18.076 764 440 VV 0.029 0.0001
23 18.117 1115 402 VV 0.043 0.0001
24 18.156 831 374 VV 0.031 0.0001
25 18.199 273 269 VV 0.017 0.0000
26 18.220 562 240 VV 0.033 0.0001
Total area = 9.5207E+008
------------------------------------------------------------------------------
std3 - second run:
where did the peaks go?

======================================
Area Percent Report
======================================
Data File Name : C:\HPCHEM\1\DATA\NV-F0138.D
Operator : Page Number : 1
Instrument : ANALYZER Vial Number :
Sample Name : Injection Number :
Run Time Bar Code: Sequence Line :
Acquired on : 16 Aug 07 07:27 PM Instrument Method: 6584-100.MTH
Report Created on: 16 Aug 07 07:59 PM Analysis Method : 6584-100.MTH
Sig. 1 in C:\HPCHEM\1\DATA\NV-F0138.D
Pk# Ret Time Area Height Type Width Area %
|---|----------|--------------|--------------|----|---------|----------|
1 1.992 20196 504 BV 0.474 0.0042
2 2.266 4.62873E+008 8.8122E+007 VV 0.066 95.3902
3 2.339 2.22797E+007 2979198 VV 0.118 4.5915
4 8.584 8414 737 BV 0.190 0.0017
5 8.631 1578 752 VV 0.028 0.0003
6 8.694 3491 1025 VV 0.057 0.0007
7 8.811 8327 1235 VV 0.085 0.0017
8 8.858 3065 1340 VV 0.032 0.0006
9 8.924 5311 1423 VV 0.053 0.0011
10 8.966 2753 1438 VV 0.027 0.0006
11 8.988 2177 1586 VV 0.023 0.0004
12 9.006 3182 1566 VV 0.028 0.0007
13 9.074 7852 1662 VV 0.059 0.0016
14 9.173 8008 1610 VV 0.071 0.0017
15 9.247 2831 1421 VV 0.033 0.0006
16 9.266 6916 1366 VV 0.065 0.0014
17 9.380 3846 991 VV 0.065 0.0008
18 9.529 814 393 VV 0.029 0.0002
Total area = 4.85241E+008
=======================================
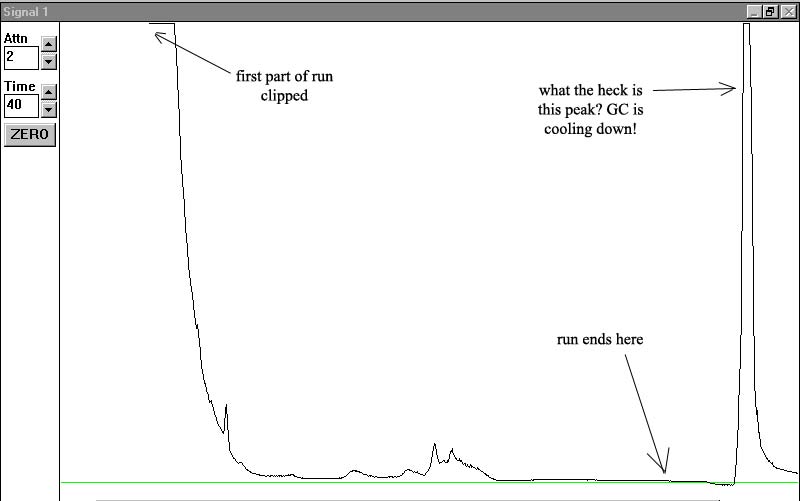
std3 - third run:
===============================================================================
Area Percent Report
===============================================================================
Data File Name : C:\HPCHEM\1\DATA\NV-F0139.D
Operator : Page Number : 1
Instrument : ANALYZER Vial Number :
Sample Name : Injection Number :
Run Time Bar Code: Sequence Line :
Acquired on : 16 Aug 07 08:09 PM Instrument Method: 6584-100.MTH
Report Created on: 16 Aug 07 08:41 PM Analysis Method : 6584-100.MTH
Sig. 1 in C:\HPCHEM\1\DATA\NV-F0139.D
Pk# Ret Time Area Height Type Width Area %
|---|----------|--------------|--------------|----|---------|----------|
1 1.243 9879 495 BV 0.269 0.0011
2 1.734 9.32837E+008 1.44431E+008 VV 0.081 99.8959
3 4.543 425222 13308 VV 0.383 0.0455
4 5.264 216745 6668 VV 0.389 0.0232
5 6.099 137964 6176 VV 0.290 0.0148
6 6.865 27234 1132 VV 0.301 0.0029
7 13.038 8469 338 PV 0.327 0.0009
8 13.128 752 252 VV 0.049 0.0001
9 13.181 1227 215 VB 0.095 0.0001
10 15.982 8772 386 BV 0.279 0.0009
11 16.159 259 171 VV 0.025 0.0000
12 16.215 568 123 VB 0.062 0.0001
13 17.413 53900 2480 BV 0.259 0.0058
14 17.797 6859 905 VV 0.101 0.0007
15 17.872 5444 1018 VV 0.082 0.0006
16 17.954 3941 958 VV 0.054 0.0004
17 18.022 3132 878 VV 0.048 0.0003
18 18.349 42734 1814 VV 0.287 0.0046
19 18.755 9241 956 VV 0.121 0.0010
20 18.848 7974 725 VV 0.183 0.0009
21 19.176 977 277 VV 0.059 0.0001
22 19.345 456 223 VV 0.033 0.0000
Total area = 9.33808E+008
==================================
I'm not sure what causes the lack of repeatability, all of our injections from day 1 last week showed pretty sharp peaks. I wonder if injection technique is to blame.
for all but the last run i drew 1 uL into the syringe and injected it. for the last run above run, I drew in 5 uL, and injected it from 5 down to 4 uL. in all cases i washed the syringe with at least three full 5uL shots of pure heptane immediately after injecting.
I guess my major questions are:
1) any clue as to why am i seeing such lack of repeatability?
2) looking at the 'good' chromatograph above, would you say the column seems to be functional at least?
3) how can i reduce the solvent tailing? replace the guard column and/or clip a meter off the front of the actual column?
4) last night i 'baked' my column at 350C for an hour (sorry bruce!, i started right before your post, and i figured 350C isnt too hot). over the baking time, the high temperature baseline decreased pretty steadily. This morning i replaced the injector septa, and then checked my carrier gas flowrate. The flowrate had substantially decreased. Before we were using 6-8 psi on the EPC to get 3 mL/min carrier gas flow. I had to bump it up to over 14 psi to get 3 mL/ minute this morning! Does that tell us anything about the condition of the column?
5) what is the odd peak in the last chromatagraph above. This is well after the run is over when the GC is cooling down. Again, does it tell us anything about the condition of the column?
thanks everybody for all your advice!
the y-axis is about 40000 in the following chromatographs
std3 - first run:
def seeing some peaks, but the solvent tail is still making it difficult.
=========================================
Area Percent Report
=========================================
Data File Name : C:\HPCHEM\1\DATA\NV-F0137.D
Operator : Page Number : 1
Instrument : ANALYZER Vial Number :
Sample Name : Injection Number :
Run Time Bar Code: Sequence Line :
Acquired on : 16 Aug 07 06:44 PM Instrument Method: 6584-100.MTH
Report Created on: 16 Aug 07 07:16 PM Analysis Method : 6584-100.MTH
Sig. 1 in C:\HPCHEM\1\DATA\NV-F0137.D
Pk# Ret Time Area Height Type Width Area %
|---|----------|--------------|--------------|----|---------|----------|
1 1.345 2606 165 BV 0.198 0.0003
2 1.567 86 17 VB 0.063 0.0000
3 1.746 7818 5836 BV 0.022 0.0008
4 1.949 8.99766E+008 1.31258E+008 VV 0.085 94.5063
5 2.004 5.0963E+007 5806131 VV 0.106 5.3529
6 4.094 962430 22965 VV 0.496 0.1011
7 5.645 230977 14254 VV 0.199 0.0243
8 9.458 41810 4701 VV 0.148 0.0044
9 9.525 19902 5259 VV 0.063 0.0021
10 9.541 59716 5182 VV 0.192 0.0063
11 17.090 1754 235 BV 0.093 0.0002
12 17.151 1039 265 VV 0.055 0.0001
13 17.214 621 238 VV 0.034 0.0001
14 17.328 1642 244 VV 0.087 0.0002
15 17.358 198 202 VV 0.016 0.0000
16 17.458 1061 170 VV 0.103 0.0001
17 17.704 1371 94 VV 0.184 0.0001
18 17.765 429 139 VV 0.040 0.0000
19 17.805 328 181 VV 0.030 0.0000
20 17.980 2333 350 VV 0.083 0.0002
21 18.043 1287 406 VV 0.053 0.0001
22 18.076 764 440 VV 0.029 0.0001
23 18.117 1115 402 VV 0.043 0.0001
24 18.156 831 374 VV 0.031 0.0001
25 18.199 273 269 VV 0.017 0.0000
26 18.220 562 240 VV 0.033 0.0001
Total area = 9.5207E+008
------------------------------------------------------------------------------
std3 - second run:
where did the peaks go?
======================================
Area Percent Report
======================================
Data File Name : C:\HPCHEM\1\DATA\NV-F0138.D
Operator : Page Number : 1
Instrument : ANALYZER Vial Number :
Sample Name : Injection Number :
Run Time Bar Code: Sequence Line :
Acquired on : 16 Aug 07 07:27 PM Instrument Method: 6584-100.MTH
Report Created on: 16 Aug 07 07:59 PM Analysis Method : 6584-100.MTH
Sig. 1 in C:\HPCHEM\1\DATA\NV-F0138.D
Pk# Ret Time Area Height Type Width Area %
|---|----------|--------------|--------------|----|---------|----------|
1 1.992 20196 504 BV 0.474 0.0042
2 2.266 4.62873E+008 8.8122E+007 VV 0.066 95.3902
3 2.339 2.22797E+007 2979198 VV 0.118 4.5915
4 8.584 8414 737 BV 0.190 0.0017
5 8.631 1578 752 VV 0.028 0.0003
6 8.694 3491 1025 VV 0.057 0.0007
7 8.811 8327 1235 VV 0.085 0.0017
8 8.858 3065 1340 VV 0.032 0.0006
9 8.924 5311 1423 VV 0.053 0.0011
10 8.966 2753 1438 VV 0.027 0.0006
11 8.988 2177 1586 VV 0.023 0.0004
12 9.006 3182 1566 VV 0.028 0.0007
13 9.074 7852 1662 VV 0.059 0.0016
14 9.173 8008 1610 VV 0.071 0.0017
15 9.247 2831 1421 VV 0.033 0.0006
16 9.266 6916 1366 VV 0.065 0.0014
17 9.380 3846 991 VV 0.065 0.0008
18 9.529 814 393 VV 0.029 0.0002
Total area = 4.85241E+008
=======================================
std3 - third run:
===============================================================================
Area Percent Report
===============================================================================
Data File Name : C:\HPCHEM\1\DATA\NV-F0139.D
Operator : Page Number : 1
Instrument : ANALYZER Vial Number :
Sample Name : Injection Number :
Run Time Bar Code: Sequence Line :
Acquired on : 16 Aug 07 08:09 PM Instrument Method: 6584-100.MTH
Report Created on: 16 Aug 07 08:41 PM Analysis Method : 6584-100.MTH
Sig. 1 in C:\HPCHEM\1\DATA\NV-F0139.D
Pk# Ret Time Area Height Type Width Area %
|---|----------|--------------|--------------|----|---------|----------|
1 1.243 9879 495 BV 0.269 0.0011
2 1.734 9.32837E+008 1.44431E+008 VV 0.081 99.8959
3 4.543 425222 13308 VV 0.383 0.0455
4 5.264 216745 6668 VV 0.389 0.0232
5 6.099 137964 6176 VV 0.290 0.0148
6 6.865 27234 1132 VV 0.301 0.0029
7 13.038 8469 338 PV 0.327 0.0009
8 13.128 752 252 VV 0.049 0.0001
9 13.181 1227 215 VB 0.095 0.0001
10 15.982 8772 386 BV 0.279 0.0009
11 16.159 259 171 VV 0.025 0.0000
12 16.215 568 123 VB 0.062 0.0001
13 17.413 53900 2480 BV 0.259 0.0058
14 17.797 6859 905 VV 0.101 0.0007
15 17.872 5444 1018 VV 0.082 0.0006
16 17.954 3941 958 VV 0.054 0.0004
17 18.022 3132 878 VV 0.048 0.0003
18 18.349 42734 1814 VV 0.287 0.0046
19 18.755 9241 956 VV 0.121 0.0010
20 18.848 7974 725 VV 0.183 0.0009
21 19.176 977 277 VV 0.059 0.0001
22 19.345 456 223 VV 0.033 0.0000
Total area = 9.33808E+008
==================================
I'm not sure what causes the lack of repeatability, all of our injections from day 1 last week showed pretty sharp peaks. I wonder if injection technique is to blame.
for all but the last run i drew 1 uL into the syringe and injected it. for the last run above run, I drew in 5 uL, and injected it from 5 down to 4 uL. in all cases i washed the syringe with at least three full 5uL shots of pure heptane immediately after injecting.
I guess my major questions are:
1) any clue as to why am i seeing such lack of repeatability?
2) looking at the 'good' chromatograph above, would you say the column seems to be functional at least?
3) how can i reduce the solvent tailing? replace the guard column and/or clip a meter off the front of the actual column?
4) last night i 'baked' my column at 350C for an hour (sorry bruce!, i started right before your post, and i figured 350C isnt too hot). over the baking time, the high temperature baseline decreased pretty steadily. This morning i replaced the injector septa, and then checked my carrier gas flowrate. The flowrate had substantially decreased. Before we were using 6-8 psi on the EPC to get 3 mL/min carrier gas flow. I had to bump it up to over 14 psi to get 3 mL/ minute this morning! Does that tell us anything about the condition of the column?
5) what is the odd peak in the last chromatagraph above. This is well after the run is over when the GC is cooling down. Again, does it tell us anything about the condition of the column?
thanks everybody for all your advice!