-
- Posts: 2846
- Joined: Mon Aug 30, 2004 7:17 am
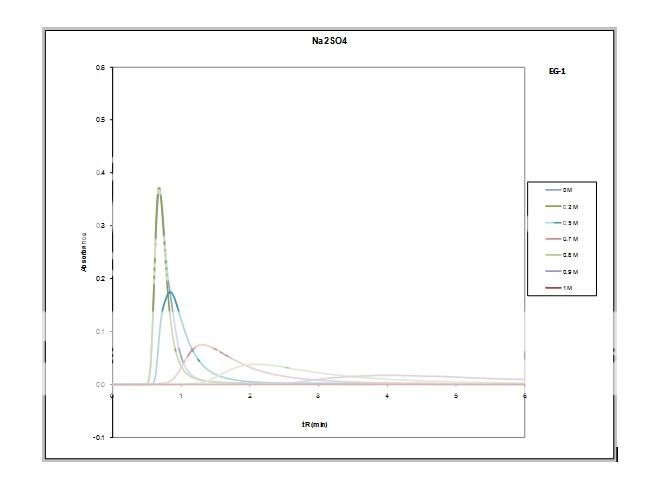
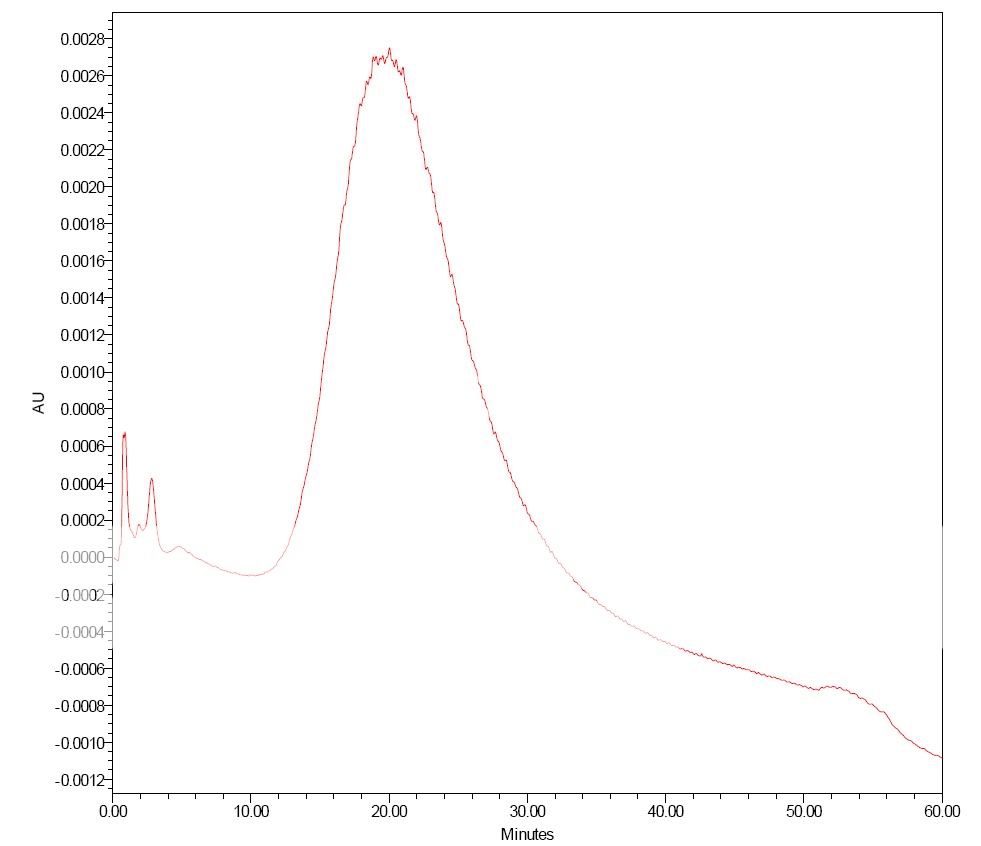
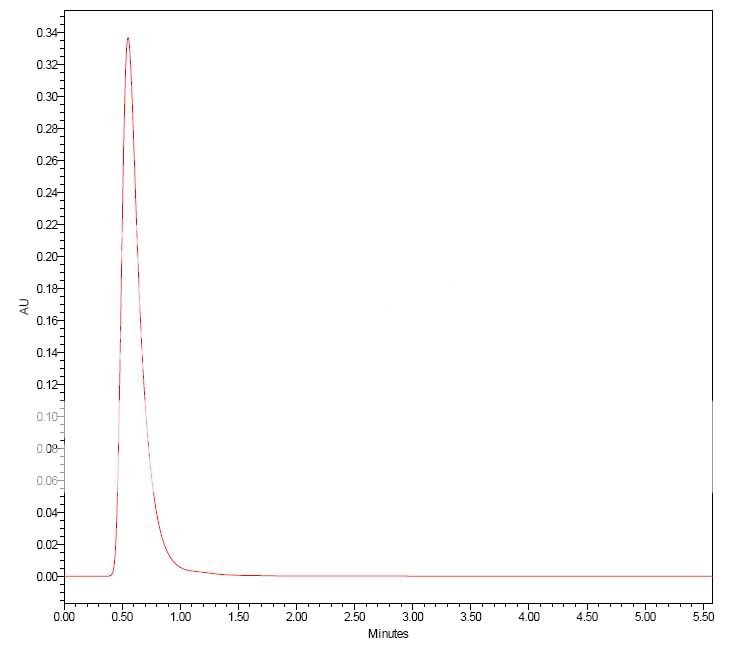
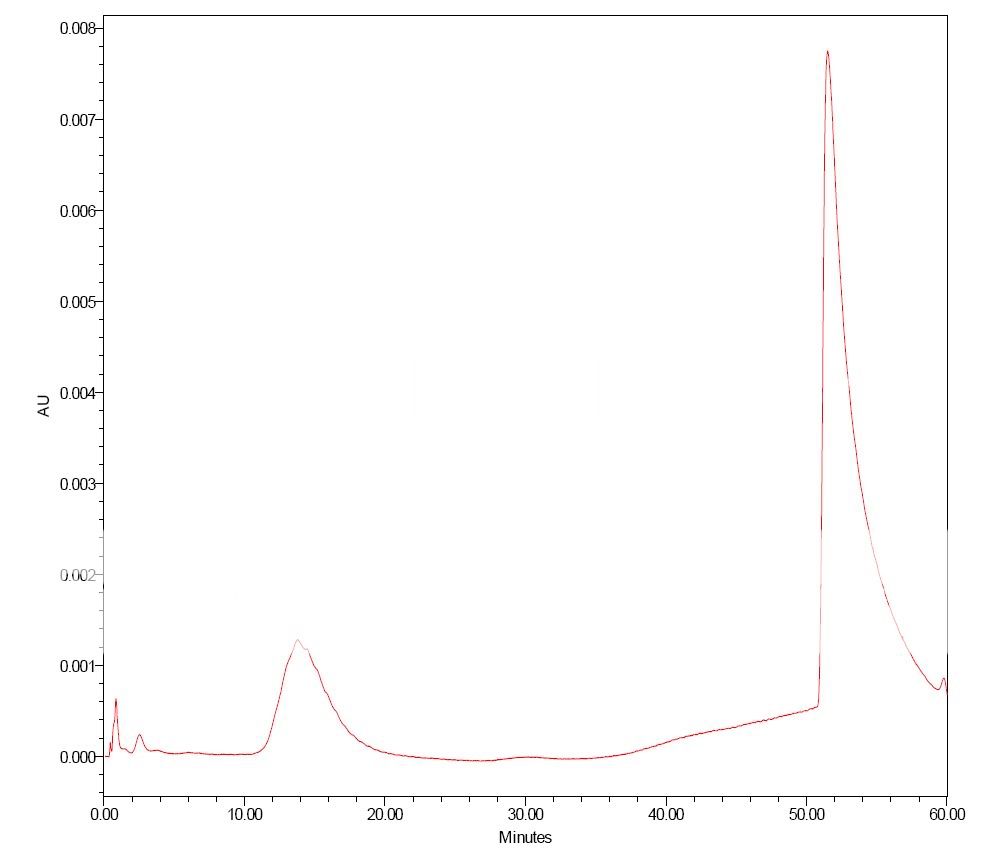
Dancho, some of these mismatches were done under extreme conditions to learn more on these phenomena. Thus some mobile phases were very low concentrations of NaOH and H2SO4. Even 0.5 µL injections could give exreme results. However, I was also surprised by some "misbehavior" with more "normal" mobile phases and inj. volumes around 5µL.
qrys, injecting the protein in 0.8M salt didn´t improve things? If this is correct, and also the protein is out of the column before the salt concentration reaches 0.2M, one runs out of explanations. Thus I also tend to go in the direction of questioning the column "chemistry".
I have precipitated proteins (mostly albumin) with PEG, also how stable is this PEG coating?
qrys, injecting the protein in 0.8M salt didn´t improve things? If this is correct, and also the protein is out of the column before the salt concentration reaches 0.2M, one runs out of explanations. Thus I also tend to go in the direction of questioning the column "chemistry".
I have precipitated proteins (mostly albumin) with PEG, also how stable is this PEG coating?